Research
Areas of focus in the Zhao Group.
01
DFT Methods for Relativistic Effects and Strong Correlations
Materials containing heavy elements exhibit numerous novel properties, placing them at the forefront of physics and chemistry research. Compared to light elements, heavy atoms in the last few rows of the periodic table have much more complex electronic structures, posing a long-standing challenge for ab-initio calculations — typically arising from relativistic effects (scalar-relativistic and spin-orbit coupling) and strong correlation effects associated with localized d/f shells.
In previous work we developed a quasi-four-component (Q4C) relativistic energy-band theory for extended systems with hundreds of atoms per unit cell, enabling accurate fully-relativistic all-electron calculations at lower computational cost. We are now developing fully-relativistic DFT+U methods and fully-relativistic hybrid functionals to address relativistic and strong-correlation effects synergistically.

02
Large-Scale DFT Code Development
We are active developers of the FHI-aims electronic-structure code — an all-electron, full-potential DFT code designed for large-scale parallel calculations. Our in-house developments are implemented under the FHI-aims framework using numerical atomic orbitals (NAO) as basis sets.
03
Computational Chemistry and Materials Science
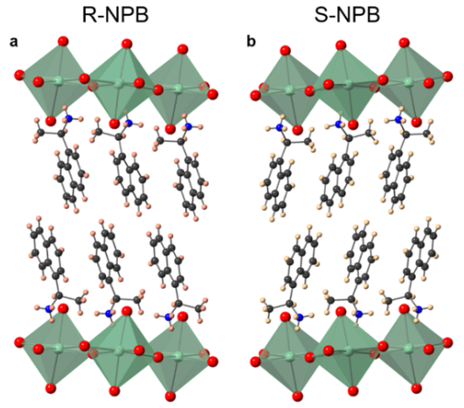
We apply the methods above to materials containing heavy elements, including perovskites, nuclear fuels, and magnetic materials.
04
Machine Learning and Quantum Computation for Electronic Structures
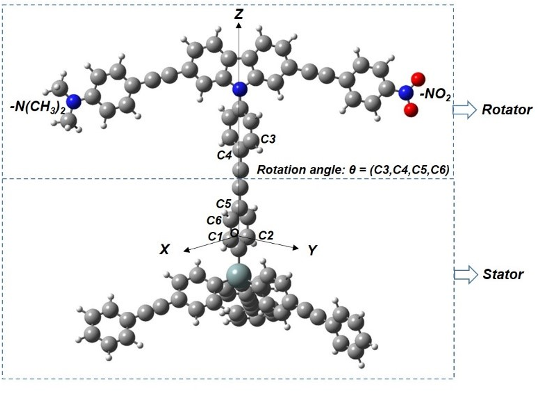
Through deep learning with convolutional neural networks on large datasets, we aim to reveal the influence of structural information — bond lengths and angles — on the magnitude of spin-orbit splittings in perovskite systems. We also develop ML-based ab-initio methods to accelerate electronic-structure calculations.
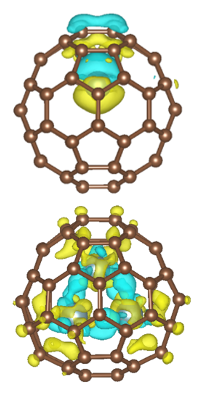
A newer direction is quantum computation. We are collaborating with teams from the Institute of Software, Chinese Academy of Sciences, and from USTC on near-term quantum computers, focusing on ab-initio algorithms and early-stage applications to quantum chemistry.